1. THALASSEMIAS
- Quantitative abnormality of globin chain synthesis
- Genetic mutation in thalassemia results in
- Absence of mRNA production from the involved gene
- Production of non-functional mRNA
- Production of an unstable mRNA that is prematurely degraded
- The above results in decreased synthesis of the involved globin chain. This has 2 consequences
- Decreased Hb synth – resulting in anaemia and microcytosis
- Aggregation of excess free globin chains – produced by the non-thalassemic gene. These aggregates of unpaired globin chains attach to and damage er cell membrane, causing hemolysis
- In severe cases, many erythroid precursors are destroyed in the BM, those that escape are prematurely destroyed by macrophages in spleen and liver
- Extremely heterogeneous
- High prevalence of thalassemia in areas with endemic malaria – Africa, Mediterranean, Middle East, India
Complications
- Chronic anaemia – leads to growth retardation, delayed sexual maturation, cardiac dilation, CHF
- Expansion of BM – Due to erythroid hyperplasia
- ‘Hair on end’ appearance on radiograph due to widening of diploic spaces in skull – EMH
- Frontal bossing of forehead. Prominent cheeks due to hypertrophy of maxillae (chipmunk appearance).
- Extramedullary haematopoiesis causes enlargement of spleen and liver.
- – chronic hyper-absorption of iron by GIT, due to chronic erythropoiesis
- Fe deposition in the heart causes cardiomyopathy and arrhythmias
- Deposition in liver causes portal fibrosis and cirrhosis (↑risk of HCC)
- Chronic hemolysis – causes splenomegaly, hepatomegaly, bilirubin gallstones
α-Thalassemia
Epidemiology
- Mediterranean, Middle East, China, SE Asia, Africa
- Single gene mutation MC in Africa
Pathophysiology
- Deletion of α globin chains
- There are 2 genes for the α globin chain on chromosome 16
| Single-gene mutation
Silent |
(-a/aa) | Asymptomatic, without microcytosis or anemia |
| 2 gene mutation
a-thal minor or trait |
(-a/-a)
(–/aa) |
Mild microcytic anemia, serious complications are rare |
| 3-gene mutation
HbH disease(β4) |
(–/-a) | Moderately severe, microcytic anemia. Excess β chains precipitate as β4 tetramers (HbH). Pts may/may not have splenomegaly, Fe overload, skeletal complications |
| 4-gene mutations
Hb Barts (γ4) |
(–/–) | Hydrops fetalis. Incompatible with life. Pregnancies terminate spontaneously. Infants who survive have severe anasarca and die of CHF. |
β-Thalassemia
Epidemiology
- Mediterranean – esp Greece and Italy (βo). Africa (β+)
Pathophysiology
- There is a single gene for β globin chain on chromosome 11
- Mutations can result in either
- A complete lack of β chain synthesis (βo-thalassemia)
- A decrease in β chain synthesis (β+-thalassemia)
| Β- Thalassemia minor | (β/β+) | Heterozygosity results in mild clinical syndrome. Mild decrease in Hb and MCV. Few sx |
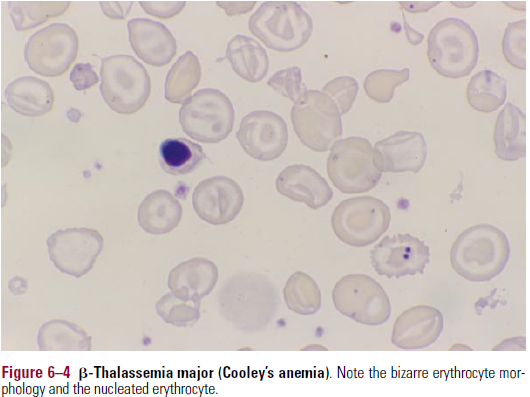
| B-Thalassemia major
Cooley’s Anemia |
(β0/β0) | Severe anemia and v.low MCV. Near total absence of HbA. Ineffective erythropoiesis, expansion of BM with skeletal complications. Splenomegaly, Fe overload due to hyperabsorption of iron |
| B-Thalassemia intermedia | (β+/β+) or (β+/β0) | Some HbA is produced |
Diagnosis
HbA – α2β2
HbA2 – α2δ2
HbF – α2γ2
HbS – sickle
- A microcytic anemia that is not due to iron deficiency is most likely thalassemia
- Blood smear – microcytosis, hypochromia
- Severe cases – anisocytosis, bizarre poikilocytes, polychromasia, nucleated er
- B-Thal diagnosed by Hb electrophoresis
- Shows increased HbA2
- Also slight increase in HbF
- A-Thal diagnosed
- A microcytic anaemia not due to IDA and has a normal level of HbA2 is most likely A-Thal
- HbH disease can be diagnosed by presence of HbH on electrophoresis (it is the fastest migrating Hb)
Treatment
- RBC transfusion – 1-3 units every 3 weeks
- Complications – alloimmunisation, Fe overlaod, infections (esp viral hepatitis)
- In pts with severe thalassemia, aim to keep Hb>12g/dL to prevent skeletal complications by shutting of erythropoein-driven erythroid hyperplasia
- Iron chelation – to treat iron overload
- Deferoxamine (Desferal) – subcutaneous infusions. Deferasirax (oral)
- Mobilises Fe so it’s excreted in urine
- Complications – cataracts, hearing loss
- Splenectomy
- Patients should be immunised against S.pneumoniae, H.influenza, N.meningitidis prior